|
|
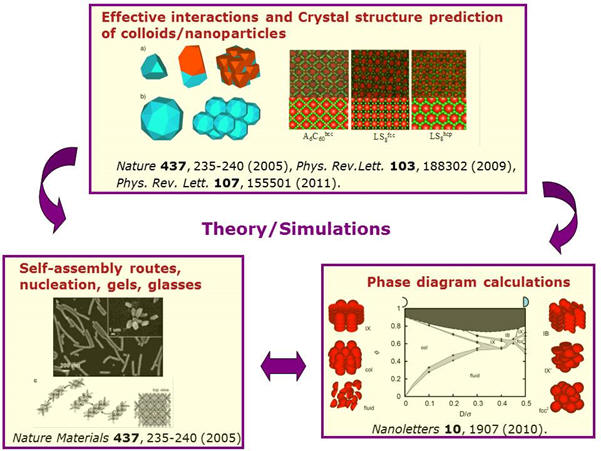
Computer Simulation
group of Marjolein Dijkstra
|
 |
Recent advances in the chemical synthesis and fabrication of colloidal
particles have resulted in a spectacular variety of new colloidal building
blocks. The main challenge is to exploit this huge variety of available
colloidal building blocks and to self-assemble them into structured
arrangements for advanced and functional materials and devices. The
fabrication of these so-called ‘nanomaterials’ with a well-defined
structure on the scale of tens to hundreds of nanometers, makes these
materials perfectly suited for the manipulation of (visible) light. Hence,
colloidal crystals with lattice spacings similar to the wavelength of
light are considered to be prime candidates for the fabrication of
photonic band-gap materials with potential applications in highly
efficient light-emitting diodes (LEDs), solar cells, sensors, and optical
computer chips. Additionally, nanomaterials with the right properties are
likely to be instrumental in the development of new photovoltaic cells and
electronic displays. The potential use of the spontaneous
self-organisation of colloids as a promising and inherent cheap route for
the fabrication of nanostructures requires not only the ability to tune
the properties of the colloidal building blocks, but also a better
understanding of the relation between the building blocks, their
interactions, and the self-assembled structures. Additionally, the tunability
of the effective interactions between the colloidal particles offers great
opportunities. To be more specific, colloidal particles with anisotropic
interactions can be synthesized by controlling the shape of the particles, or
by creating ‘patches’ on the surface of the particles. The interactions
between the particles can also be altered by modifying the dispersive medium, i.e.
the addition of salt to the dispersion leads to screening of the electrostatic
interactions, the presence of non-adsorbing polymer results in effective depletion
attractions, critical Casimir forces arise due to the confinement of
long-range density fluctuations when the host fluid is close to a critical point, and
solvent-mediated interactions can arise when the solvent approaches a binodal. One
can further modify the interactions by application of e.g. external electric and
magnetic fields, templates, gravity, etc. Exploiting the self-assembly of these
novel colloidal building blocks calls for theoretical tools to predict the structure
and phase behavior of these particles.
Our research group uses theory and simulations to obtain a better fundamental
understanding on how colloidal building blocks self-assemble and how the self-assembly
process can be manipulated by external fields such as gravity, templates, air-liquid
or liquid-liquid interfaces, and electric fields. In order to predict the
structural properties and phase behavior of colloidal particles, one first
identifies the possible ‘candidate’ structures in which the particles with
given interaction potentials and system parameters may assemble. To this
end, we have developed an efficient and robust method based on simulated
annealing to predict candidate structures of colloidal building blocks. The
equilibrium phase diagram can then be determined by calculating free
energies of the candidate structures in Monte Carlo simulations. In
addition, it is important to investigate the kinetic pathways and nucleation
rates to form the equilibrium structures as the self-assembly may be suppressed by kinetic
effects such as vitrification, gelation, defects and stacking faults. A new promising
research direction is to employ so-called active systems which continuously convert
energy either from an internal energy source, e.g. catalytic reactions, or from an
external source, like electric or magnetic fields, into motion to obtain structures
with new properties and functionalities that cannot be achieved in equilibrium, and
may perform tasks on demand and respond to external stimuli. We employ simulated annealing
techniques to predict candidate structures, and Monte Carlo,
(event driven) Molecular and Brownian Dynamics simulations, Stochastic Rotational Dynamics
simulations to include hydrodynamics, Umbrella and Forward flux sampling,
free-energy calculations based on thermodynamic integration methods, to determine
the (non)-equilibrium phase behavior of colloids, nanoparticles, liquid
crystals, etc. A better insight in the self-assembly process is essential
for developing new materials.
Current Research Projects
Phase behavior of experimentally realizable polyhedral colloidal particles
Anjan Prasad Gantapara, a.p.gantapara@uu.nl, phone: 030-2532467
Monte Carlo simulations, Einstein integration, variable box-shape NPT simulations
Self-assembly is ubiquitous in nature. The shape of the basic building block plays a
key role in determining the structure of the self-assembly. Motivated by recent breakthroughs
in the synthesis of colloidal polyhedral particles and their experimental application, we
study the phase behavior of faceted particles using simulations which are experimentally
available. We apply Monte Carlo simulations and free-energy calculations to determine the
phase behavior of (I) a family of colloidal truncated hard cubes in three dimensions and
(II) bi-frustums adhering to a two-dimensional air-toluene interface. For the truncated
cubes, we find a remarkable diversity in crystal phases and close-packed structures depending
sensitively on the particle shape [1]. For the bi-frustums we employ theoretical free-energy
calculations to determine the equilibrium configuration of these particles at the interface
and use this as input for simulations to determine the phase behavior. The phase behavior of
these bi-frustums adhered to the air-toluene interface matches the experimental observations [2]
as shown below. Our results illustrate the intricate relation between phase behavior and
building-block shape and can guide future experimental studies on polyhedral-shaped nanoparticles.
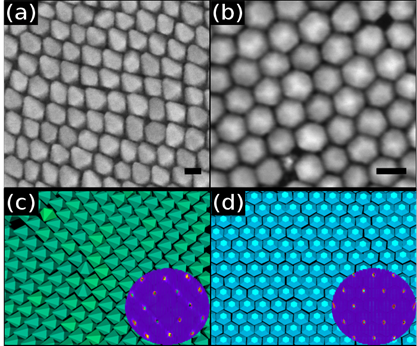
Figure:
(a) and (b) are SEM images of self-assembled superlattices of hexagonal bipyramid-shaped and hexagonal
bifrustum-shaped ZnS nanoparticles respectively. Snapshots of the self-assembled
superlattices
of hexagonal bipyramids (d,e) and hexagonal bifrustums (f) obtained from
Monte Carlo simulations. Corresponding diffraction patterns are displayed
in the insets.
[1] W. van der Stam, A.P. Gantapara et al., Nano Letters 14, 1032 (2014)
[2] A.P. Gantapara et al., Phys. Rev. Lett. 111, 015501 (2013)
The origin of chirality in colloidal liquid crystals
Simone Dussi, s.dussi@uu.nl, phone: 030-2532467
Monte Carlo simulations, molecular dynamics simulations,
Einstein integration, Onsager theory, density functional theory (DFT)
Chirality plays a paramount role in life, chemistry, and materials
science. A chiral molecule lacks an internal plane of symmetry and has a
non-superimposable mirror image. In life, many of the biologically active
molecules are chiral (e.g. most amino acids are left-handed, whereas sugars
are right-handed) and these building blocks can self-assemble in
higher-ordered structures (e.g. helix protein structures, DNA). Chirality is important in the formation of biological ordered structures but also in
the field of colloidal liquid crystals.
Whereas the nematic director of a common (uniaxial) nematic
liquid-crystal phase is homogeneous throughout the system, the cholesteric
phase displays an helical arrangement of the director field. In spite of the
huge relevance for optoelectronic applications, the relation between the
macroscopic chirality of the liquid crystalline phase and the microscopic
chiral details (shape, interactions) of the constituent molecules is still
not well understood.
The overall aim of this project is indeed to investigate the microscopic
origin of the macroscopic chirality in colloidal liquid crystals using new
theoretical approaches and state-of-the-art computer simulations. The first
objective is to study whether colloidal hard particles with a chiral shape
(e.g. hard helices) can form a purely entropy-driven cholesteric phase.
Suspensions of sterically stabilized fd virus particles exhibit a cholesteric phase [1], that might be driven by
entropy alone.
Such a mechanism was already proposed by Straley [2] in the
context of the elastic theory of liquid crystals. Extending Straley's
approach we developed a second-virial density functional theory that allows
to determine the equilibrium pitch of selected hard chiral particles [3].
Interesting scenarios are obtained varying the microscopic details and the
packing fraction of the system.
By means of ad-hoc computer simulations techniques we are able to
investigate the cholesteric phase and to check to what extend the
theoretical predictions are valid.
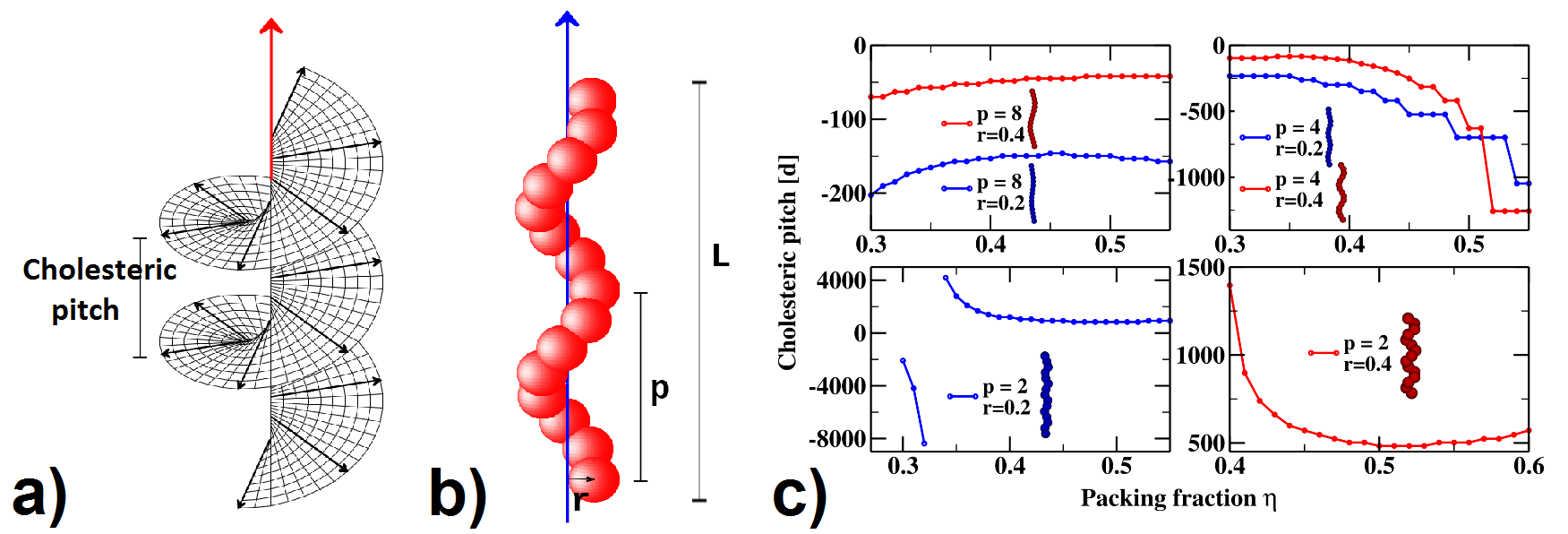
Figure:
(a) Schematic representation of the chiral nematic (a.k.a. cholesteric) phase.
(b) Example of modeling a chiral hard particle.
(c) Cholesteric pitch as a function of packing fraction for different values of
the microscopic parameters [3].
[1] E. Grelet and S. Fraden, Phys. Rev. Lett. 90, 198302 (2003)
[2] J.P. Straley, Phys. Rev. A 14, 1835-1841 (1976)
[3] S. Belli and S. Dussi et al., arXiv:1404.2113
Self-assembly of patchy colloidal particles
Guido Avvisati, g.avvisati@uu.nl, phone: 030-2538176
Monte Carlo simulations, molecular dynamics simulations
The spontaneous formation of patterns and structures is an ubiquitous
phenomenon in our world, spanning multiple length and time scales: galaxies
found in our universe are probably the most widely known example of such a
pattern formation, but to this subject belong also, for example, lane
formation in pedestrian dynamics and vortices in hydrodynamics. The
spontaneous auto-organization process of nano-to-micrometer colloidal
particles into supra-molecular equilibrium structures is referred to as
colloidal self-assembly [1] and is another example of emergent behaviour.
Not only does this process poses many fundamental and interesting physics
questions, but it also provides us with a cheap route to smart material
synthesis and therefore has the potential to hit the industrial world,
eventually our everyday life, with larger impact. One way to guide the
self-assembly is to engineer colloidal particles with discrete, attractive
interaction sites (patches) at prescribed locations on the surface of the
particles [2].
We investigate the self-assembly scenarios of patchy particles by means of
computer simulations. By using the Monte Carlo technique we ask ourselves
what is the influence of particle geometry and particle-particle interaction
on the resulting self-assembled structures. In the following picture, we can
see three examples of structures formed by attractive hard dumbbells which
we use as a model for rough-smooth colloidal dumbbells with surface
roughness specific depletion interactions [3].
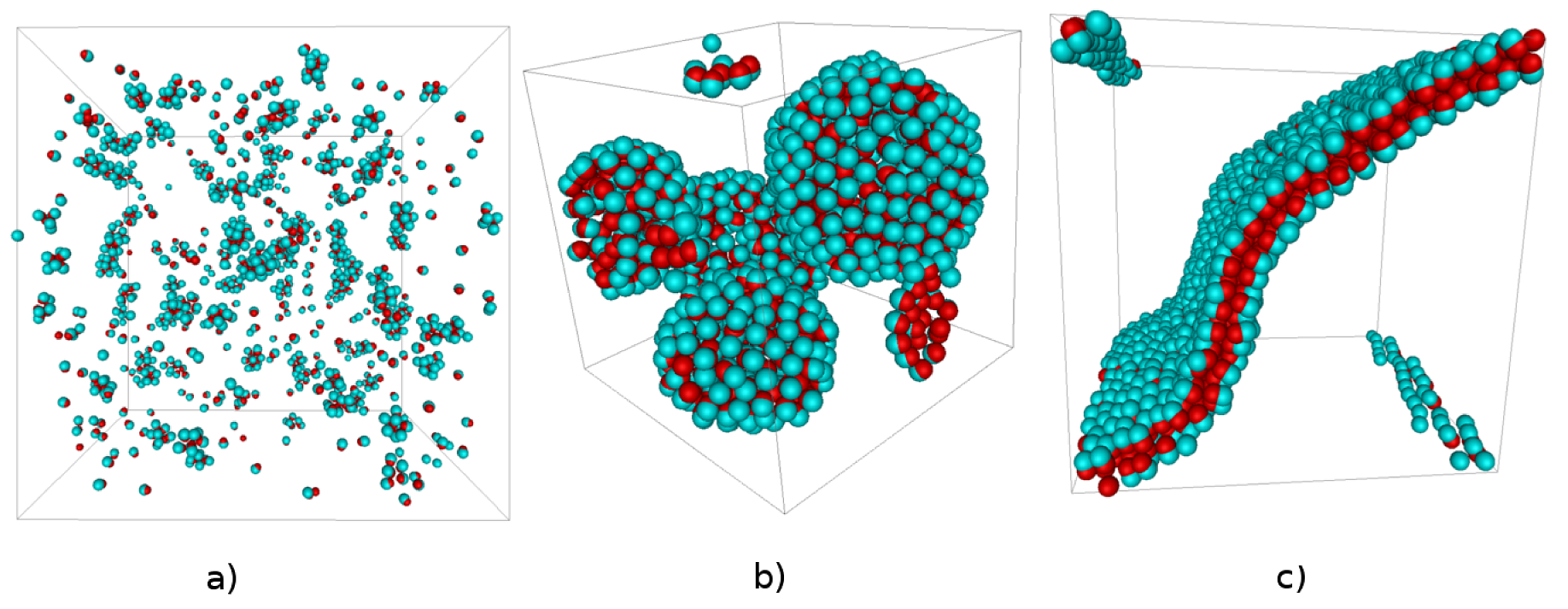
Figure:
Self-assembly products of attractive hard dumbbells: (a) Micelles,
(b) Vesicles, (c) Bilayers.
[1] G.M. Whitesides et al., Science 295, 2418-2421 (2002)
[2] S.C. Glotzer et al., Nature Materials 6, 557-562 (2007)
[3] D.J. Kraft et al., PNAS 109, 10787-10792 (2012)
Towards colloidal self-assembly mediated by near critical solvents
Nick Tasios, n.tasios@uu.nl, phone: 030-2538176
Monte Carlo simulations
Colloidal self-assembly provides means for efficient and inexpensive
fabrication of new, complex materials and devices. The self-assembly is driven by the effective interactions of
the colloids; being able to tune these interactions would, therefore, give us control over the self-assembly
process. Solvent-mediated (SM) interactions, such as the critical Casimir effect (first predicted by Fisher
and de Gennes [1]) and wetting induced interactions, are generally thought to drive particle
aggregation and colloidal self-assembly processes in a tunable, reversible, and in-situ fashion due to
their strong dependence on temperature and on solvent composition. Understanding these interactions is, thus,
critical in paving a path to tunable colloidal self-assembly.
For this project, we have developed a coarse-grained model of a
colloid-solvent mixture for which we perform Monte Carlo simulations.
Effective interactions and the phase diagram of the colloids immersed in a
near critical solvent are determined using canonical ensemble and Transition
Matrix Monte Carlo [2] simulations for neutral colloids, with equal affinity
for both solvent species, as well as for colloids which preferentially
adsorb one of the solvent species [3].
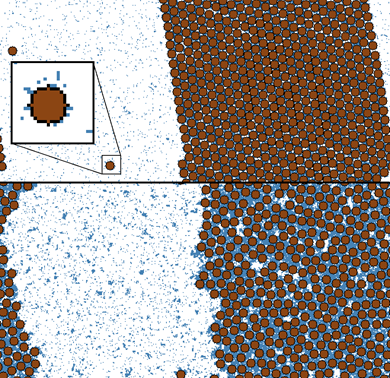
Figure:
Snapshots of canonical Monte Carlo simulations of a solvent-solvent-colloid mixture.
The snapshots show colloidal liquid-gas and solid-gas coexistence. At the top,
a region has been magnified. Blue and white pixels represent the solvent
species, while a sphere is depicted at the center[3].
[1] M. Fischer et al., C. R. Acad. Sci. B 287, 207-209 (1978)
[2] J.R. Errington, J. Chem. Phys. 118, 9915-9925 (2003)
[3] J.R. Edison, N. Tasios et al., arXiv:
Quasicrystalline structure in soft matter
Harini Pattabhiraman, h.pattabhiraman@uu.nl, phone: 030-2538176
Monte Carlo simulations
The aim of this work is to study the formation of quasicrystals in colloidal
systems. Quasicrystals are ordered, non-periodic structures initially
discovered in metallic systems. Recently, various quasicrystalline phases
have been observed in soft matter systems like binary nanoparticle mixtures,
terblock star polymers, and micellar systems [1].
Theoretical studies have confirmed the presence of photonic band gaps (PBGs)
in quasicrystals, which prevent the propagation of light of certain
wavelengths. In addition, by virtue of the quasicrystalline structure, these
PBGs are found to be less sensitive to structural defects [2]. Colloidal quasicrystals,
of sizes in the range of visible light, can result in solar cells with
increased photon path length, thereby, increasing the chance of photon absorption.
Further, the arrangement of nanoparticles can serve as a plasmonic structure
characterized by strong Mie resonance, which can be used to enhance the efficiency
of light-trapping in solar cells.
The current project deals with a study of quasicrystalline phases in colloidal
systems using Monte Carlo simulations. The challenge is to stabilize a
quasicrystal in a system
of submicron-sized colloidal particles in order to obtain non-periodic
structural correlations on a length scale close to that of visible light. The systems
studied experimentally so far are much smaller in particle size, and are ranging from 0.5 nm
metallic alloys to 80 nm star polymers. Initially, a quasicrystalline
system of binary disks, to model a binary nanoparticle mixture, will be
simulated. The PBG structure and the Mie scattering of this system will then
be calculated.
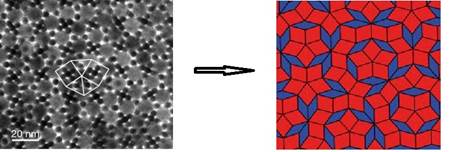
Figure:
Arrangement of particles in a binary mixture that form a quasiperiodic
structure
[1] T. Dotera, Israel J. Chem. Phys. 51, 1197-1205 (2011)
[2] K. Ueda et al., Phys. Rev. B 75, 95122 (2007)
Solvent mediated interactions between colloids in near critical solvents
John R Edison, j.r.edison@uu.nl
Monte Carlo simulations, density functional theory (DFT)
A binary mixture close to its critical point (consolute point) experiences
large scale fluctuations in composition, characteristic of a second order
phase transition. If a single colloidal particle preferring one of the
species of the binary solvent is introduced in the mixture, thick films of
preferred solvent species preferentially adsorb on its surface. The film
thickness is determined by the correlation length of the solvent, a quantity
that diverges upon approaching the critical point. When adsorbed films of
two different colloids interact they give rise to an effective long-ranged
force, with universal scaling properties. Fisher and de Gennes [1] first
predicted the presence of these long-ranged forces. These near-critical
solvent mediated (SM) interactions are also termed as critical Casimir
forces due to an analogy with the quantum Casimir effect.
The critical Casimir force can be attractive or repulsive depending on the
nature of the interactions between the colloidal surface and the solvent
species. For two identical colloidal particles in a near-critical solvent
the force between them is attractive. If the two colloids (or a wall and
colloid) are different such that they prefer different solvent species the
force is usually repulsive. The range of these forces depends on the range
of solvent fluctuations, i.e. the correlation length. Hence by tuning the
proximity to the system's critical point one can possibly drive the colloids
to aggregate or re-disperse.
Our objective is to estimate the solvent-mediated interaction between two
colloidal particles at a series of near critical thermodynamic states of the
solvent. There are several serious challenges involved in simulating such a
system. To begin with the size of the colloid to solvent should be at least
a factor of ten and naturally they have very different relaxation time scales.
Moreover close to the critical point of the solvent, critical slowing down
occurs, and large system sizes are required for a meaningful description of the
system. Hence we have begun our investigation by describing the colloid solvent system
with a coarse grained lattice based model [2]. We further plan to add ions to
the solvent to study the effects of ion-solvent coupling upon
solvent-mediated interactions.

Figure:
Two colloids of size D suspended in a near-critical solvent
whose correlation length is
ξ.
[1] M. Fisher et al., C. R. Acad. Sci. B 287, 207-209 (1978)
[2] J.R. Edison, N. Tasios et al., arXiv:
Self-assembly of patchy particles
Zdenek Preisler, z.preisler@uu.nl, phone: 030-2532320
Monte Carlo simulations, free-energy calculations, variable
box-shape NPT simulations
During the last few years the particle anisotropy, including both, an
anisotropy in interaction as well as an anisotropy in shape, has been
recognized as a gateway towards new functional materials, attracting a lot
of interest from various fields including physics and chemistry. One of the
fundamental ideas motivating many of the current studies is based on
self-assembly of building blocks into supra-colloidal structures. Instead of
a difficult and complex fabrication of a given material, one would simply
let building blocks, in our case colloids, assemble into a particular
structure by themselves [1]. Furthermore, the applicability of this idea has
been already demonstrated experimentally by S. Granick using particles with
two attractive patches, which self-assemble into a 2D kagome lattice [2].
Later the same result was also obtained using computer simulations [3].
Following the above mentioned concept, we will study the phase behavior of
patchy particles by means of computer simulations and free-energy
calculations of both ordered as well as disordered phases. We will
focus, in particular, on the investigation of a system of twelve-patch
particles and a system composed of a mixture of particles with one to twelve
patches. Apart from equilibrium structures, we will also investigate
non-equilibrium arrested states e.g. gel structures composed of these
particles.
|
|
|
|
|
 |
|
 |
|
Figure:
(left) Snapshot of a single 12-patch particle modeled using the
Kern-Frenkel potential. The interaction volume is depicted in transparent
blue and the particle hard-core is depicted in red; (right) Snapshot of a
system of the 12-patch particles.
[1] S. C. Glotzer, et al., Nature Mater. 6, 557-562 (2007)
[2] Q. Chen, et al., Nature 469 381-384 (2011)
[3] F. Romano, et al., Soft Matter 7, 5799-5804 (2011)
|